
L’ictère néonatal
Dominique Debray, Unité d’Hépatologie pédiatrique, hôpital Necker-Enfants malades, Paris
L’ictère est le symptôme le plus fréquent observé en période néonatale. La bilirubine plasmatique est physiologiquement non conjuguée et liée à l’albumine. Elle est conjuguée dans le foie par une glucuronide transférase (UGT1A1). La bilirubine conjuguée (BC), hydrosoluble est ensuite sécrétée dans la bile grâce à des transporteurs. Les causes d’ictère néonatal sont nombreuses, mais il importe d’identifier précocement une atrophie des voies biliaires.
L’ictère à bilirubine non conjuguée (BNC)
C’est l’ictère le plus fréquent (99 % des cas). Il est caractérisé par l’absence de décoloration des selles et des urines de couleur normale.• L’ictère simple du nouveau-né (NN) à terme concerne deux tiers des NN. Il est en général modéré (hyperbiliubinémie libre < 250 µmol/l), apparaît vers le 2e jour de vie et régresse avant 10 jours de vie.• Cependant, un ictère à BNC peut dans certaines circonstances apparaître plus précocement, être intense et se prolonger au-delà de J10.Les causes en sont nombreuses :– prématurité. L’ictère régresse en général avant J21 ;– hémolyse : notamment incompatibilité érythrocytaire (Rh, AO) ; hémoglobinopathies ; déficits enzymatiques érythrocytaires (G6PD, PK) ;– infections ;– résorption d’hématomes ;– diminution de l’activité de l’ UGT1A1 : lait de mère ; hypothyroïdie congénitale ; cause toxique, médicamenteuse ou infectieuse ; syndrome de Gilbert (mais se manifeste le plus souvent à l’adolescence ou à l’âge adulte) ; maladie de Crigler-Najjar type 1 et 2, très rare (incidence 1/1 000 000 naissances vivantes).L’hyperbilirubinémie libre, surtout lorsqu’elle est associée à des cofacteurs de morbidité, peut présenter une toxicité neurologique, dont la forme la plus sévère est l’ictère nucléaire. Ce risque neurologique justifie les efforts de dépistage et de prévention, fondés sur la mesure transcutanée systématique de la BNC dès J1 (avec dosage sanguin si la BNC transcutanée est élevée), la prise en compte des facteurs de risques et le traitement précoce par photothérapie. Des recommandations ont été émises quand à l’indication de la photothérapie tenant compte du terme, des facteurs de risques associés et de la valeur de la BNC(1).
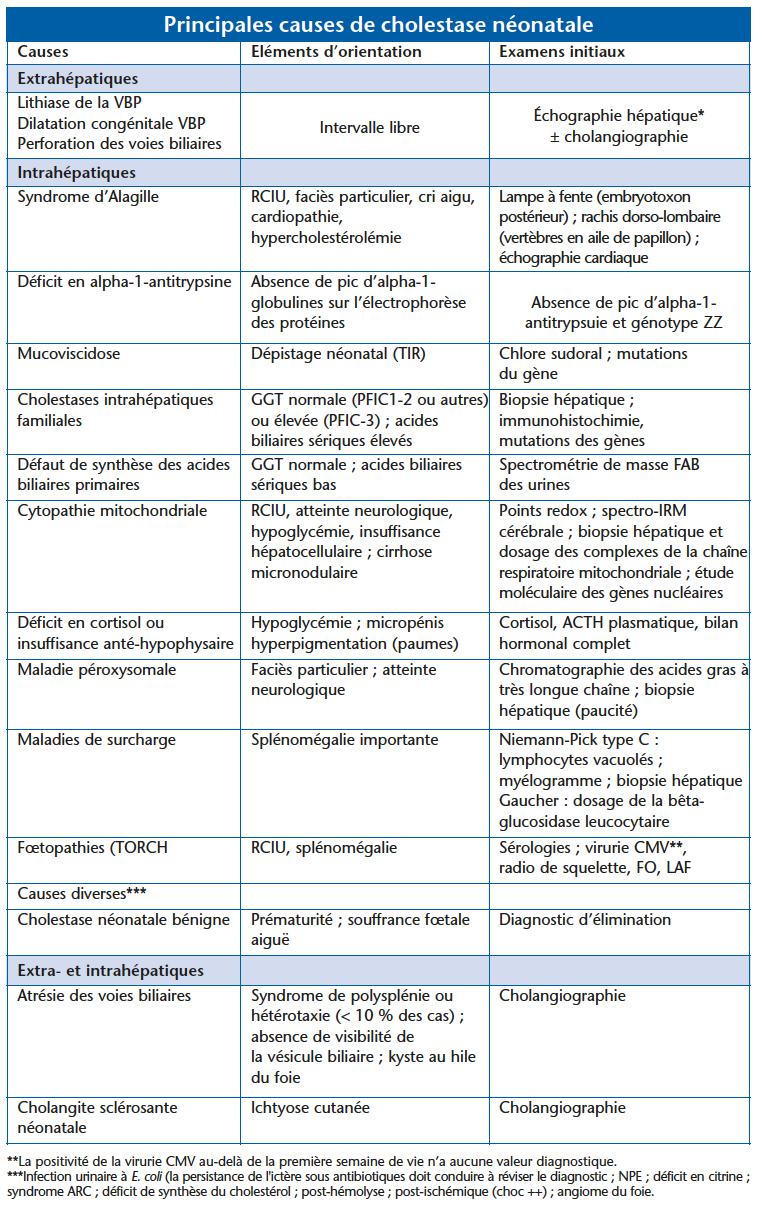
L’ictère néonatal à BC (cholestatique)
Il est beaucoup plus rare. Mais il est urgent de reconnaître sa nature cholestatique, d’en préciser la cause et d’orienter le NN vers un service spécialisé ou la prise en charge thérapeutique peut être une urgence. Il est recommandé de réaliser un dosage plasmatique de la bilirubine totale et de la BC chez tout NN présentant un ictère se prolongeant au-delà de 15 jours (même en cas d’allaitement maternel)(2). L’éducation des mères à la sortie de maternité concernant la nécessité de consulter en cas de persistance de l’ictère au-delà de 15 jours ou en cas de décoloration des selles est un point important, qui est désormais noté dans le carnet de santé.• L’ictère à BC est exceptionnellement lié à une anomalie du transport canaliculaire de la bilirubine, sans altération du flux biliaire (syndrome de Dubin-Johnson et au syndrome de Rotor). Dans ce cas, l’examen clinique, les transaminases et l’activité GT sont normaux. Le pronostic est bon sans évolution vers la fibrose.• L’ictère à BC est le plus souvent signe de cholestase. Par opposition à l’ictère à bilirubine libre, l’ictère cholestatique s’accompagne d’urines foncées. Les selles peuvent être de coloration normale, partiellement décolorées ou totalement décolorées (figure 1). Une hépatomégalie est le plus souvent notée, dont la consistance varie en fonction de la cause de la cholestase. La confirmation de la cholestase repose sur des examens biologiques simples : hyperbilirubinémie conjuguée, augmentation des phosphatases alcalines, augmentation des transaminases en général modérée (< 10 fois la normale). L’activité sérique de la Gammel GT est souvent augmentée mais peut être normale. L’étude de l’hémostase révèle rarement des signes d’insuffisance hépatocellulaire, mais peut révéler des signes d’hypovitaminose K (baisse des cofacteurs de la coagulation FII et FVII + X) qui doit être systématiquement prévenue par l’administration de vitamine K1 par voie parentérale. Le diagnostic étiologique est orienté par l’anamnèse, l’examen clinique (couleur des selles, consistance du foie, splénomégalie, faciès particulier, atteintes extrahépatiques, etc.), des examens biologiques simples et l’échographie hépatobiliaire (figure 2). Les principales causes de cholestase néonatale figurent dans le tableau. Elles peuvent être d’origine extrahépatique, intrahépatique ou extraet intrahépatique. L’atrésie des voies biliaires [AVB] est la cause la plus fréquente. L’importance de son diagnostic précoce, dont le pronostic est lié à la précocité de l’intervention chirurgicale correctrice, doit faire hospitaliser tout nouveau-né atteint de cholestase avec selles décolorées dans un centre médicochirurgical spécialisé.
Figure 1. Cholestase néonatale. A. Coloration foncée des urines. B. Coloration variable des selles.
Figure 2. Démarche diagnostique face à une cholestase néonatale.• En cas de décoloration complète et permanente des selles En l’absence de dilatation des voies biliaires évocatrice de lithiase ou de dilatation congénitale de la voie biliaire principale (le plus souvent liée à une anomalie de jonction bilio-pancréatique) à l’échographie, de déficit en alpha-1-antitrypsine (de génotype ZZ), d’éléments évocateurs d’un syndrome d’Alagille et de mucoviscidose, il s’agit a priori d’une AVB. L’échographie peut parfois montrer des signes évocateurs d’AVB : syndrome de polysplénie (rates multiples, veine porte préduodénale, retour veineux azygos, situs inversus abdominal) ; petite vésicule biliaire à paroi épaissie et/ou irrégulière après un jeûne prolongé, ou ne se vidant pas après la prise d’un biberon ; kyste visible au niveau du hile du foie ; triangle hyperéchogène au niveau de la bifurcation portale. Une échographie normale n’exclut pas le diagnostic d’AVB. Il est urgent de programmer une laparotomie pour réaliser une cholangiographie et l’intervention correctrice si le diagnostic d’AVB est con firmé devant l’absence d’opacification des voies biliaires intrahépatiques et/ou du duodénum. Certes, l’existence d’une consanguinité, d’une récurrence dans la fratrie peuvent suggérer le diagnostic de cholangite sclérosante de début néonatal, pouvant mimer cliniquement une AVB, mais en aucun cas il ne faut déroger à l’indication de la cholangiographie.• En cas de décoloration partielle ou transitoire des selles Il s’agit le plus souvent d’une cholestase d’origine intrahépatique, dont la cause est souvent identifiée par des examens complémentaires simples (tableau). La normalité permanente des gamma-GT oriente vers le diagnostic de cholestase fibrogène familiale (PFIC-1 ou 2) ou d’un défaut de synthèse des acides biliaires primaires. Si la cause n’est pas identifiée, une biopsie hépatique à l’aiguille peut être indiquée et orienter le diagnostic. Des signes histologiques d’obstacle sur les voies biliaires (fibrose porte, néocanaux biliaires, thrombibiliaires dans les voies biliaires interlobulaires) doivent faire considérer le diagnostic d’AVB et une opacification des voies biliaires est indiquée afin d’éliminer formellement une AVB. Une paucité ductulaire (absence de voies biliaires dans la majorité des espaces portes visibles) à l’examen histologique du foie n’exclut pas le diagnostic d’AVB. Des cellules géantes (hépatocytes multinuclées) sont fréquemment notées quelle que soit l’étiologie, mais leur grand nombre en l’absence de fibrose peut orienter vers une cholestase néonatale bénigne. Si la cause n’est pas reconnue, une surveillance clinique et biologique s’impose. L’absence de recoloration franche des selles doit faire envisager une nouvelle biopsie hépatique à l’aiguille. Si la cholestase régresse, il est nécessaire de poursuivre une surveillance clinique et biologique pendant au moins 1 an pour s’assurer de la guérison complète qui confirme le diagnostic de cholestase néonatale transitoire bénigne.
Conclusion
Il est indispensable de reconnaître rapidement la cause d’un ictère chez l’enfant. Il est urgent d’en apprécier la nature : ictère à BNC ou ictère à BC. Les deux risques immédiats sont la toxicité neurologique (ictère nucléaire) en cas d’hyperbilirubinémie libre importante et l’hypovitaminose K en cas de cholestase. Le diagnostic étiologique est urgent. Les observations d’enfants atteints d’AVB diagnostiquées tardivement (≥ 2 mois de vie) alors que les signes de cholestase étaient évidents dès la période néonatale (selles blanches à la sortie de maternité) restent encore trop nombreuses.
• American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 2004 ; 114 : 297-316. • Moyer V et al. Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2004 ; 39 : 115-28.